2024年5月21日,,華中科技大學同濟醫(yī)學院基礎(chǔ)醫(yī)學院胡清華聯(lián)合華中科技大學同濟醫(yī)學院附屬同濟醫(yī)院蔣丁勝以及武漢大學人民醫(yī)院易欣團隊在心血管領(lǐng)域?qū)W術(shù)期刊Circulation Research發(fā)表題為“SMYD2-methylated PPARγ Facilitates Hypoxia-induced Pulmonary Hypertension by Activating Mitophagy”的研究論文,,首次發(fā)現(xiàn)SMYD2單甲基化非組蛋白PPARγ并抑制其核轉(zhuǎn)位和激活,通過引發(fā)線粒體自噬加速肺動脈平滑肌細胞增殖,,促進肺動脈高壓的發(fā)生發(fā)展,,且SMYD2的抑制劑LLY-507和PPARγ的激動劑Rosiglitazone均能有效逆轉(zhuǎn)缺氧誘導的肺動脈高壓,,這表明靶向抑制SMYD2或激活PPARγ是防治肺動脈高壓的潛在策略。
肺動脈高壓(pulmonary hypertension, PH)是一類致命的肺循環(huán)障礙性疾病,,通常始于原發(fā)或繼發(fā)的各種原因?qū)е碌钠骄蝿用}壓升高,,繼而引發(fā)肺循環(huán)血流受阻以及右心衰竭。異常壓力和微環(huán)境下的肺血管重構(gòu)是肺動脈高壓的典型病理特征,,肺動脈中層平滑肌細胞作為肺血管壁的主要組成部分,,其異常增殖是導致肺血管重構(gòu)的核心機制。蛋白質(zhì)甲基化是一種可逆的蛋白質(zhì)翻譯后修飾,,通常發(fā)生在組蛋白上,,調(diào)控染色質(zhì)的結(jié)構(gòu)影響基因的表達。近年來,,多個非組蛋白也被報道能發(fā)生甲基化修飾,,并影響相關(guān)蛋白的活性和功能。多項研究表明,,蛋白質(zhì)甲基化修飾參與肺動脈高壓的調(diào)控,,如組蛋白甲基轉(zhuǎn)移酶G9a和EZH2可通過調(diào)節(jié)肺動脈平滑肌細胞的凋亡、增殖和遷移,,參與肺動脈高壓的病理過程,。但蛋白質(zhì)甲基化修飾,尤其是非組蛋白的甲基化修飾在肺動脈高壓中的作用和分子機制尚不完全清楚,,且靶向蛋白質(zhì)甲基化修飾是否能有效逆轉(zhuǎn)缺氧誘導的肺動脈高壓尚缺乏相關(guān)證據(jù),。SMYD2是一種蛋白質(zhì)賴氨酸甲基轉(zhuǎn)移酶,能促進組蛋白和非組蛋白的甲基化修飾調(diào)控包括腫瘤在內(nèi)的多種疾病的發(fā)生發(fā)展,但SMYD2在肺動脈高壓中的調(diào)控作用和分子機制尚無報道,。
首先,,研究人員發(fā)現(xiàn),在3型肺動脈高壓臨床患者以及慢性缺氧誘導的肺動脈高壓模型大,、小鼠的肺動脈平滑肌中層中SMYD2的表達上調(diào),。采用LLY-507 (SMYD2抑制劑)處理雌、雄性大鼠以及小鼠可以顯著逆轉(zhuǎn)慢性缺氧誘導的肺血管重構(gòu)和右心重構(gòu),;相反,,平滑肌細胞特異性SMYD2過表達(SMYD2-vTg)小鼠則表現(xiàn)出更為嚴重的缺氧相關(guān)肺動脈高壓表型。
繼之,,研究人員對大鼠原代肺動脈平滑肌細胞(RPASMCs)進行缺氧刺激(1% O2)并分別給予LLY-507,、SMYD2敲低或過表達處理。結(jié)果顯示,,缺氧誘導RPASMCs增殖能力增強,,LLY-507或敲低SMYD2顯著抑制缺氧誘導的RPASMCs增殖;相反,,SMYD2過表達則促進缺氧誘導的PRASMCs增殖,。接著,研究團隊對敲低SMYD2后的RPASMCs進行了轉(zhuǎn)錄組學測序,。生物信息學分析結(jié)果顯示,,SMYD2對細胞周期、DNA復制等生物過程具有顯著調(diào)節(jié)作用,,且敲低SMYD2后G2/M 檢查點相關(guān)分子表達下調(diào),。后續(xù)實驗證實SMYD2可通過阻止細胞由S期進入G2/M期從而抑制PASMCs增殖。
進一步地,,通過單樣本基因組富集分析(ssGSEA),,研究人員發(fā)現(xiàn),敲低SMYD2后PPARγ信號通路活性顯著增強,。進一步研究表明,,SMYD2并不影響PPARγ的表達,而是通過與PPARγ相互作用并單甲基化PPARγ,,進而阻止PPARγ的核轉(zhuǎn)位影響其對下游基因的轉(zhuǎn)錄活性,。鑒于PPARγ對線粒體功能的重要調(diào)控作用,,研究團隊通過mt-Keima熒光探針,、透射電鏡以及Seahorse線粒體壓力測試等手段探索并證實SMYD2過表達可促進缺氧誘導的線粒體自噬、MDA/ROS釋放和線粒體能量代謝異常,,而采用PPARγ激動劑Rosiglitazone (ROSI)處理則可顯著逆轉(zhuǎn)SMYD2過表達對線粒體自噬的影響,。
最后,研究團隊對慢性缺氧誘導的SMYD2-vTg小鼠給予Rosiglitazone (ROSI)處理,結(jié)果顯示,,激活PPARγ可顯著改善SMYD2過表達導致的缺氧相關(guān)肺血管重構(gòu)和右心衰竭,;另一方面,體外實驗也證實,,ROSI處理顯著逆轉(zhuǎn)了缺氧條件下SMYD2過表達誘導的RPASMCs過度增殖,。
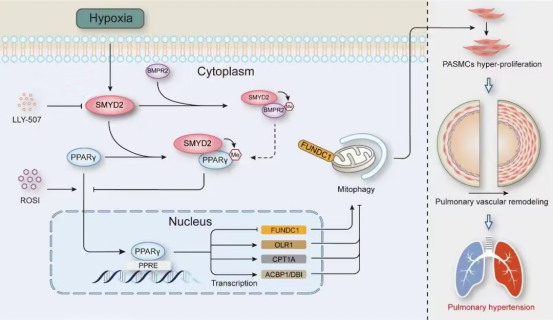
綜上所述,本研究揭示了蛋白質(zhì)甲基轉(zhuǎn)移酶SMYD2在肺動脈平滑肌細胞增殖,、肺血管重構(gòu)以及肺動脈高壓發(fā)生發(fā)展中的關(guān)鍵作用,,闡明了SMYD2對非組蛋白PPARγ的甲基化修飾、亞細胞定位和活性調(diào)控作用,,提出SMYD2-PPARγ軸通過調(diào)控線粒體自噬誘導肺動脈平滑肌細胞增殖參與肺動脈高壓形成的表觀遺傳學機制,,靶向抑制SMYD2或激活PPARγ可能成為防治肺動脈高壓的新靶點。華中科技大學同濟醫(yī)學院附屬同濟醫(yī)院心臟大血管外科李毅博士,、魏翔教授以及華中科技大學基礎(chǔ)醫(yī)學院肖瑞講師為論文的第一作者,,武漢大學人民醫(yī)院易欣副教授、華中科技大學同濟醫(yī)學院附屬同濟醫(yī)院心臟大血管外科蔣丁勝副研究員以及華中科技大學基礎(chǔ)醫(yī)學院胡清華教授為論文共同通訊作者,。該研究獲得國家自然科學基金項目資助,。
原文鏈接:https://www.ahajournals.org/doi/abs/10.1161/CIRCRESAHA.124.323698
 English
English

